Per meeting questions for the “Talk about RNA synthesis with Stanford team”
I asked jee questions about the player puzzles and modeling vs nature, A part of it was could they be sent to the lab ?
(below is parts from a chat log)
jeehyung: We could certainly look into it. However, there are few possible problems.
jeehyung: 1. They weren’t designed under lab constraints (no consecutive 4 Gs or Cs). And they didn’t have the tails required for the synthesis
Tatyana Zabanova: question… “they didn’t have the tails required for the synthesis”… What tails are required?
jeehyung: for RNA to be synthesized they need to have special sequences at the beginning & end. If you look at RNA lab, there are locked sequences at the beginning & end. Those are tails
Something I’d be curious about is how well natural RNA sequences score in lab – have they done any comparisons with similarly sized known RNA sequences?
I’d like to hear more about the tails too.
And my main questions relate to the SHAPE data – how it’s gathered and processed, what it means, how it’s used along with nearest neighbor constraints to generate the “estimated” structures we get in results. How confident they are in those results. What do they think is going on when one nucleotide appears to be definitely bonded (has very low SHAPE number) but there’s no obvious candidate for it to be bonded to (this will happen sometimes with adenine especially it seems to me, there’ll be one or two that look very much bonded, without any Uracil candidates for it to be bonded to).
I’m sure I’ll come up with more before April 18, but that seems like a good start
Some of the SHAPE data from “Shape Library 101 : The Finger” have radically different range of values from other designs in the same lab. Why are the range of SHAPE data different? What is the metric to decide correct threshold?
The ‘Energy Codon Table’ and ‘Tetraloops Table’ both show that there are certain sequences that are favoured for stability in RNA folding. Have these values been verified in the lab and are these findings born out by the natural synthesis results the team has had so far?
For example - several of Mat747’s lab designs do not use tetraloop stabilisation which would put them at a disadvantage from a ViennaRNA algorithm perspective, yet they score better in synthesis than others that do make use of stable tetraloop sequences.
Is the latter a factor of the interpretation of the SHAPE data? Does the threshold as Ding mentions above play a role?
Though I do wonder if the SHAPE data is less of a blunt evaluation instrument than using RNAfold or similar. Finding out how the threshold is determined might give some insight into how accurate or not SHAPE is.
Are there any preliminary discoveries/insights that have been gained from the lab results so far? Some theories that the synthesis team would like us as players to explore?
Other than the SHAPE data, are there other measures being used to explore the synthesis results? If yes, what and why?
Chaen
edit: to consolidate all my question posts into a single one.
What are the constraints for puzzles for the lab? The four consecutive Gs and Cs as well as the tails mentioned above are two. What others should be taken into account if some of us are thinking of designing puzzles that could potentially be synthesized at a later date?
Just wanted to chime in to reply to some of boganis’s excellent questions.
>Why synthesize RNA, what is the current view on what synthesized RNA could be used for?
RNA is a critical part of the ‘computing power’ of many viruses (including HIV & influenza), cancer cells, and normal cells. There are many labs and companies who want to design other RNAs to disrupt or take advantage of natural RNAs – but there’s a lot of trial-and-error because our current folding rules are imperfect, as you all know. In an upcoming stage of EteRNA we’ll start designing these kinds of switches and activators, and hopefully start testing them in real cells. I think this could start happening in a few months, especially as we find other labs who would be good collaborators. [By the way, I want to emphasize that everything we are doing here is in the public domain and for the benefit of science.]
>How will lab results and bot folding results be used to improve folding algorithms?
That is the immediate next stage. EteRNA will have the most impact if everyone in the world acan benefit from your collective analysis and intuition. So we want to harness player intuition into automated algorithms – like an EteRNA server – that anyone can then run online. We’re designing a set of puzzles and, hopefully, a convenient interface to crowd-source that algorithm writing and test the algorithms rigorously. You’ll be getting an announcement about this from Jee over the next month.
>Too bad that player puzzles can’t be synthesized, I would have loved to see my nick written in synthesized RNA!
Well, maybe that will happen soon… my lab just synthesized some RNAs that spell out our name when we do our capillary electrophoresis experiments! Right now, the main rate-limiting step to synthesizing all player designs – ‘choose your own adventure!’ – is the cost of the material. This cost is affordable at 8 designs/week, but would go out of our current (small) budget if we scale up further. We’ve got some innovations in the pipeline that could potentially increase the throughput massively, but I can’t promise anything concrete yet.
Boganis: I can’t answer for the eterna team, but I should point out that in the field of bioinformatics in general, algorithms are usually published and the software open-sourced so that they can enjoy as much circulation and vetting as possible. People tend be to be very skeptical of closed-source and proprietary algorithms because you can’t reproduce their results under less-than-ideal scenarios. So i doubt anyone from eterna are planning to get rich on that front, compounded with the fact that it’s very hard to patent an abstract algorithm.
Of course, the flip side of “public domain” means that it’s free for anyone else to use (think TIVO and linux), they just can’t claim they invented it. But that’s inevitable in any science that produces useful knowledge. . .
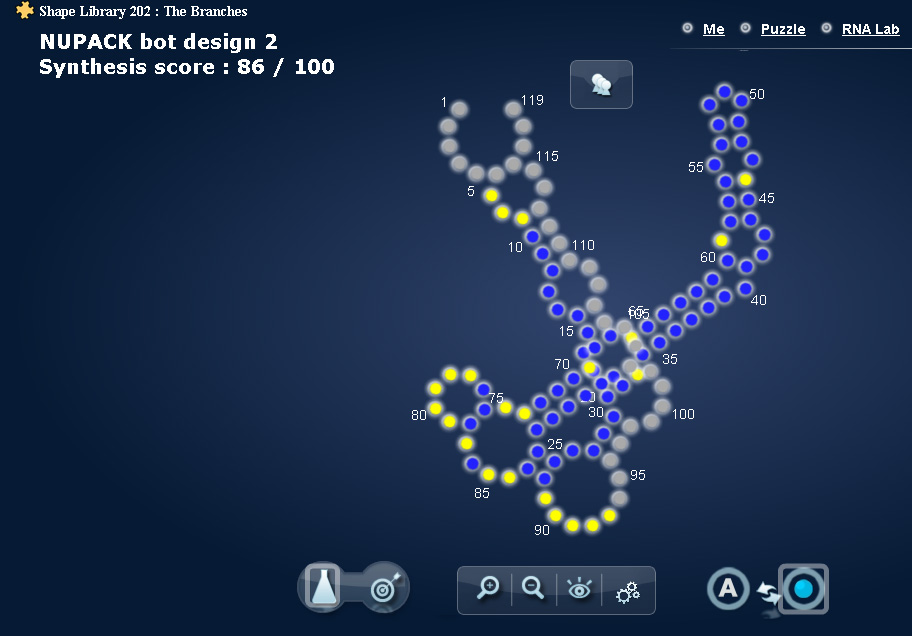
Looking at the round two bot results - NUPAK bot #2 scored a result of 86, but it appears to misfold spectacularly - yet it scores higher than other designs where at the shape holds at least partially.
It puts a question mark over the accuracy of SHAPE data as a predictor of a successful design for me, as it seems to favour any pairing for scoring, even if the shape is wildly inaccurate.
Or is the visual misfold a result of Eterna’s interpretation of the SHAPE data not reliable as a measure of the SHAPE results?
The “estimate” fold in EteRNA just try to “guess” how RNA folded based on the SHAPE data. On a tricky shape like this, the algorithm is likely to come up with a bad estimation.
In general, the “estimate” mode is less reliable than the SHAPE data.
here’s the answer – the one on the right is ‘perception series 2’, and its SHAPE data is about as good as it gets. The one on the left has something weird going on around resides 10-20, possibly an alternative structure with the U’s forming noncanonical pairs – but I don’t yet have a good model.
“Something I’d be curious about is how well natural RNA sequences score in lab – have they done any comparisons with similarly sized known RNA sequences?”
I’m enclosing plots of the SHAPE reactivity for two RNAs with known crystal structure, the P4-P6 domain of the Tetrahymena ribozyme and an adenine-sensing riboswitch. You can find the known 3D structures here and here. They get eterna scores of 89 and 98 respectively. For some other RNAs (not shown), we get EteRNA scores ranging from ~83 up to 98.
These are both widely studied ‘model systems’ in RNA folding. The lines mark the min/threshold/max values that are automatically set in getting the EteRNA score, and the red curve marks which positions should be unpaired or paired given the known crystal structure. Green x’s mark positions where the data aren’t within the range considered good in EteRNA scoring. Most of these spots correspond positions at the very edges of helices, and some correspond to positions that are ‘unpaired’ in secondary structure but actually form noncanonical (‘tertiary’) base pairs in the full 3D fold of the RNA.
It would be wonderful for us to find an experimental way to take these natural RNAs of known fold and get EteRNA scores of 100 when we get the experimental data; and to take incorrect folds and assign them bad scores. The problem right now is that the empirical SHAPE-based ‘calls’ on whether a residue is paired or unpaired is great, but can get thrown off by fluctuations at the edges of helices or by noncanonical interactions. That means that some of your designs may fold up ‘correctly’ in the test tube, but the SHAPE data miscalls them as incorrect – for example, the loops look ‘protected’ to SHAPE but are actually just fine. Unfortunately we have no quick empirical way to verify this; my lab is developing better methods (see the 2D mapping paper on our lab website ), but these require more resources, and we can only apply them to a few designs per year. So for the weekly EteRNA cycle, we are asking players to meet a ‘higher standard’ where the SHAPE data needs to be as perfect as possible. We’re trying to work on more sensitive and accurate ways to validate or falsify designs. For now if you get ~83 or above you might be right – work hard to get above 96!