Introduction
A complex RNA secondary structure can be viewed as a collection of simpler substructures. The goal of this strategy is to use past synthesis data to build a library of sequence fragments organized by the substructures they were intended to create. The information contained within this library then can be used to build or rank new sequences for structures that have not been investigated in lab.
Sequence fragments should not be re-used if they have failed to form the desired substructure in past syntheses. Conversely, sequence fragments should be re-used if they frequently permit the desired substructure to form.
Identifying a substructure
The previous lab, “Water Strider,” will be used as an example.
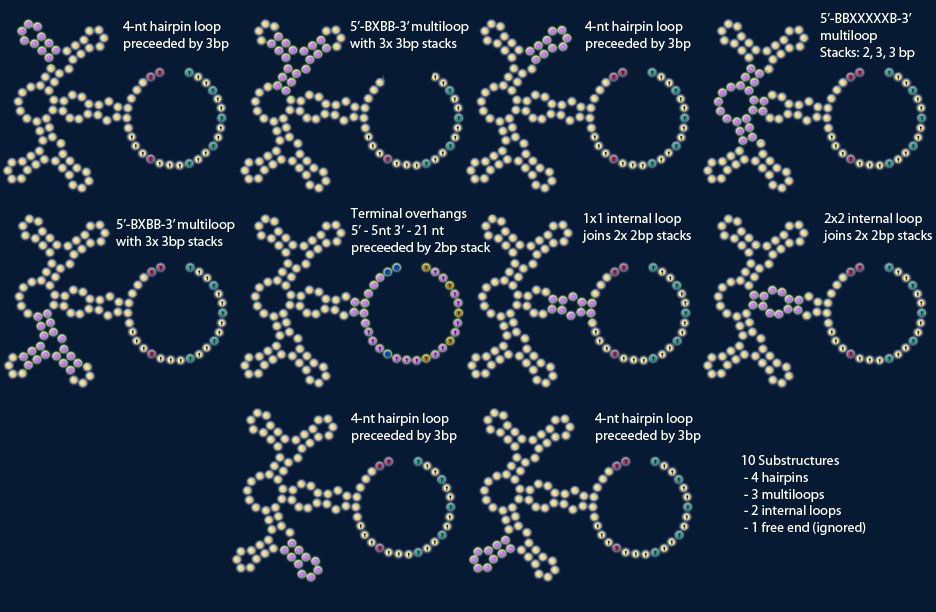
Each loop (hairpin, internal, or multi-branched) has a corresponding substructure that consists of the loop and its adjoining stacks. “Fused loops” should be counted as a single entity with one corresponding substructure. The sequences for all stacks would appear twice in the fragment library. This is not necessarily problematic, however.
A note on my loop nomenclature: Starting from branch closest to the 5’ end of the molecule, go around the loop and list whether there is a branch (B) or unpaired base (X). You’ll end up with something like 5’-BXBB-3’ or whatever.
Describing a substructure
What information should be specified about a substructure? I envision a hierarchical organization of the fragment library, from general to specific information about the substructure.
- All sequence fragments
– Basic substructure types (hairpin loops, internal loops, multiloops, fused loops)
— General substructure information (loop sizes, branch distribution in multiloops)
---- Moderately specific information (length of stacks attached to loops)
----- Very specific information (instances in molecule)
Each sequence fragment would also be described by two other pieces of information: the number of times it has been synthesized, and the number of times it has successfully folded into the intended substructure.
A simple example
Things obviously get more convoluted when multiloops, asymmetric internal loops, and fused loops enter the picture. Also, there is still the matter of how less-than-exact matches are used when ranking or building new sequences. I’ll be posting my ideas in the next few days, but any and all input is welcome.